|
Back to 2014 Annual Meeting Posters
PTK6 Increases Apoptosis With Gemcitabine Treatment in Pancreatic Cancer Cells by Enhancing DNA Damage
Hiroaki Ono*, Marc D. Basson, Hiromichi Ito
Surgery, Michigan State University, East Lansing, MI
Background:
Protein Tyrosine Kinase 6 (PTK6) is a non-receptor type tyrosine kinase known to be aberrantly expressed in various cancers including pancreatic cancer. The role of PTK6 in cancer chemo-resistance remains unknown. We tested our hypothesis that PTK6 regulates gemcitabine (GEM) resistance in pancreatic cancer, and explored its mechanism.
Methods:
We studied 2 human pancreatic cancer cell lines, Panc1 and MIAPaCa2. For some studies, GEM resistant clones of Panc1 and MIAPaCa2 were isolated by culture with GEM for 2 months. Cell survival was measured by WST-8 assay. GEM-induced apoptosis and DNA damage were evaluated by Western blotting. The effect of PTK6 overexpression on the efficacy of GEM therapy for pancreatic cancer in vivo was assayed using a xenograft mouse model.
Results:
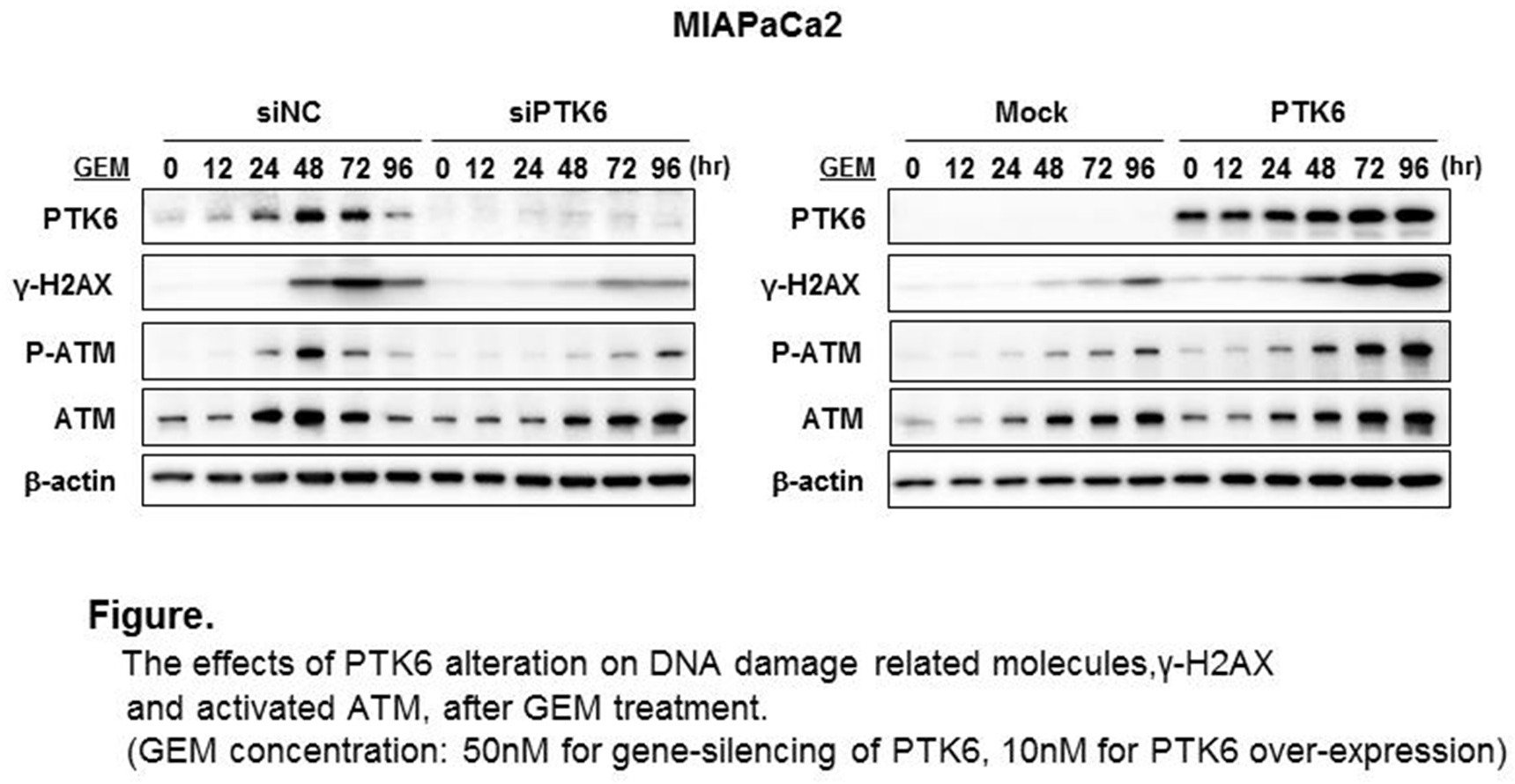
Endogenous PTK6 expression was increased at 24-48 hours with GEM treatment in Panc1 and MIAPaCa2 cell lines. PTK6 gene-silencing increased cell survival after GEM treatment and decreased cleaved Caspase3 and PARP, indicating decreased apoptosis, while PTK6 overexpression decreased cell survival and increased apoptosis. Basal PTK6 expression was significantly reduced in the GEM resistant clones compared with the parental lines (0.29 fold decrease in MIAPaCa2 and 0.60 fold decrease in Panc1, respectively, p<0.05). Restoration of PTK6 expression using an over-expression vector made the resistant MIAPaCa2 clone cells sensitive to GEM (0.77 fold decreased survival compared to the resistant clone, p<0.05). To explore the mechanism in which PTK6 regulates the cytotoxic effect of GEM on pancreatic cancers, we tested the effect of altered PTK6 expression on DNA damage induced by GEM. GEM-induced H2AX phosphorylation (γ-H2AX), which is a specific marker for DNA double-strand breaks, was significantly reduced by PTK6 gene-silencing, while GEM-induced γ-H2AX was increased by PTK6 overexpression. Furthermore, PTK6 gene silencing also inhibited the GEM-induced activation of ataxia-telangiectasia mutated (ATM) protein kinase, a central initiator of key signal responses to DNA damages in both cell lines. Conversely, ATM kinase activation was enhanced by PTK6 overexpression (Figure). In the mouse xenograft model, subcutaneous tumors with PTK6-overexpressing MIAPaCa2 showed significant growth reduction compared with tumors with control MIAPaCa2 after 5 weeks treatment with GEM (150 mg/kg IP, twice a week) (606 mm3 vs 252 mm3 in size, 609 mg vs 128 mg in weight, p<0.01, respectively) .
Conclusion:
PTK6 is endogenously activated by GEM treatment in pancreatic cancers, and forced-overexpression of PTK6 increases the efficacy of GEM on pancreatic cancer with enhancing DNA damage. Further study to elucidate the mechanism by which PTK6 regulates GEM-induced DNA damage may identify therapeutic targets to improve the outcomes of patients with pancreatic cancer.
 The effects of PTK6 alteration on DNA damage related molecules,γ-H2AX and activated ATM after GEM treatment for MIAPaCa2 cells: left panel, PTK6 was gene-silenced with siRNA and cells were treated with 50nM of GEM, right panel, PTK6 was over-expressed and cells were treated with 10nM of GEM.
Back to 2014 Annual Meeting Posters
|